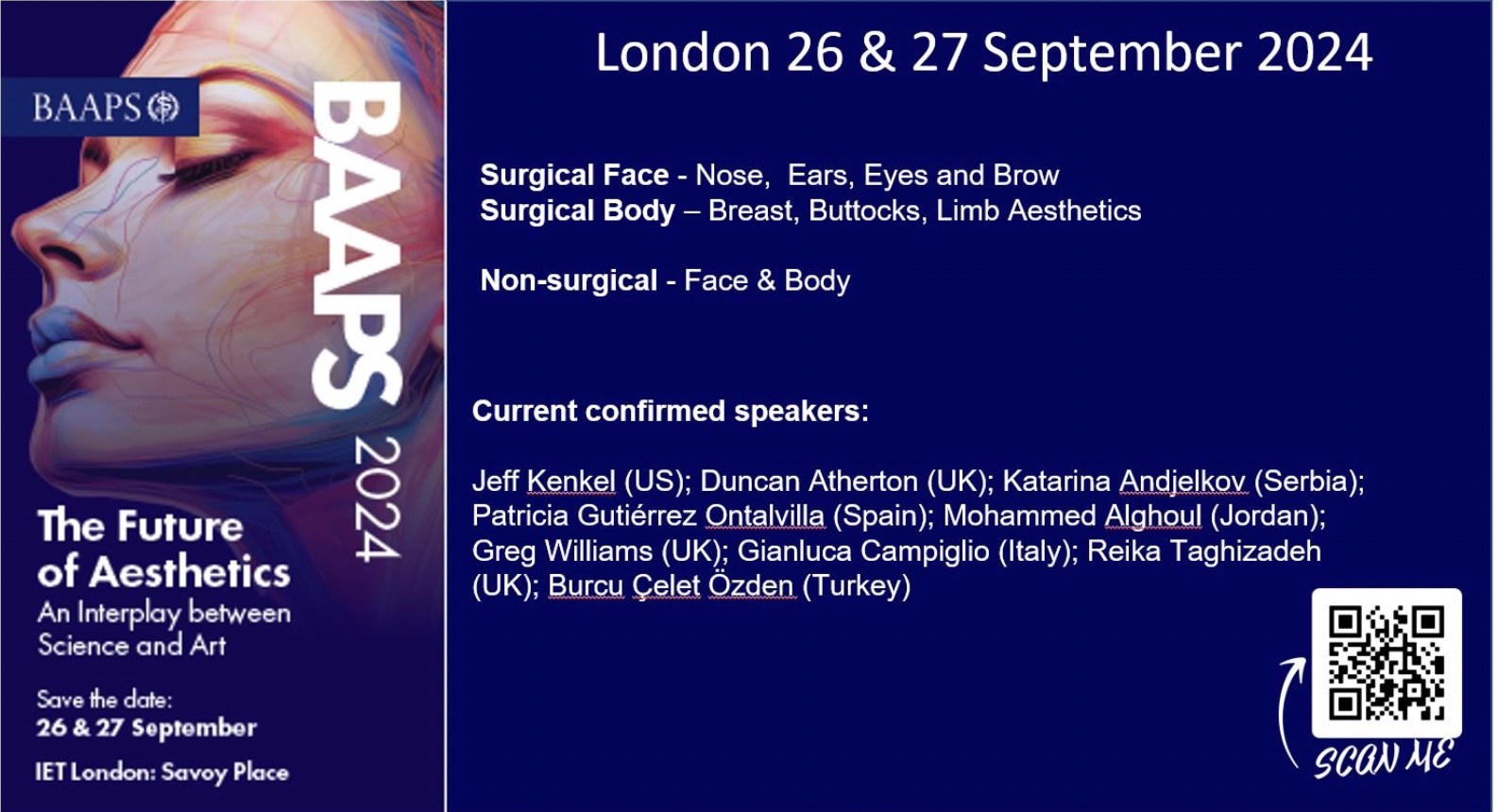
A word from Birgit Stark
ESAPS President
Dear colleagues,
Dr. Carlos Parreira kindly handed the governing responsibilities of ESAPS/EASAPS over to me
after approval by the general assembly in Lisbon 2022. To serve European aesthetic societies
(EASAPS) as well as individual plastic surgeons (ESAPS) is a great honor but also a
challenge. Together with past president Ivar van Heijningen, immediate past president Carlos
Parreira and president elect Michel Rouif, we discussed and defined the future path for
ESAPS/EASAPS some years ago and our thoughts are well documented in the “long term vision”
document. The next years to come will therefore follow a logical and sequential pathway.
Dr. Carlos Parreira kindly handed the governing responsibilities of ESAPS/EASAPS over to me
after approval by the general assembly in Lisbon 2022. To serve European aesthetic societies
(EASAPS) as well as individual plastic surgeons (ESAPS) is a great honor but also a
challenge. Together with past president Ivar van Heijningen, immediate past president Carlos
Parreira and president elect Michel Rouif, we discussed and defined the future path for
ESAPS/EASAPS some years ago and our thoughts are well documented in the “long term vision”
document. The next years to come will therefore follow a logical and sequential pathway.
During my term 2022 -2024, I will humbly serve to promote our core values so well defined by
dedicated and hardworking earlier leaders and individuals of our society. Together with my
team, we will focus on creating value for individual members and societies by promoting
Aesthetic Plastic and Reconstructive Surgery as one united speciality. In my view, we should
bridge the gap between academic reconstructive plastic surgery and aesthetic surgery for the
benefit of patients. Understanding and mutual respect for both medical fields can help to
improve patients’ outcomes by improved systematic and comprehensive education of young
colleagues. It is therefore a pleasure to work on standards for Aesthetic Plastic Surgery
training and present regular educational events in Aesthetic Plastic Surgery. Preparation of
the scientific programme of the next biennial meeting in Gothenburg, April 24 has already
been undertaken.
In addition, we offer the excellent “Patient Safety Handbook” edited by Dr. Jesús Benito
Ruiz and his team and are preparing a database for relevant scientific publications. Updates
on Public Hearings, CEN communications, Medical Device Regulations and interactions with
media will be communicated in our newsletters and presidential retreats. Internally, we have
much work to do to improve the ESAPS administrative structure and define an internal code of
conduct with a background of clear governance definitions and structure. We are expecting
interesting discussions and regular interactions with our industry partners with a common
endpoint of promoting and improving patient safety.
My primary function will be to orchestrate all committees within a democratic environment
and spirit to end up with a well performing symphony without dissonances. I value the input
and contributions of all aesthetic plastic surgeons in Europe. ESAPS and EASAPS have no
future without your valuable confidence in my intentions and aspirations and those of our
fantastic colleagues in different committees serving our specialty of Aesthetic Plastic
Surgery.
Expand